The N+1 Problem
The N+1 problem in genomics refers to the inefficiency of interrogating existing samples for read coverage when a new sample introduces novel variants into a cohort. The gVCF format addresses this problem by introducing ranges that distinguish reference from no-calls. However, the gVCF format alone does not solve the broader performance problem when adding new samples, which has parallels to a similar problem in databases: adding a new row with a field that doesn’t exist as a column in the database. That would require adding a new column for that field, and filling null values for all existing rows.
More specifically, from a data management perspective, the N+1 problem is introducing the following severe performance issues in existing solutions:
- The new sample needs to alter the entire existing dataset, typically stored in a CombinedVCF file (similar to adding a column in the above database example). This impacts storage size and write performance.
- Or, the new sample must be stored separately, implying that all samples are stored as single-sample (g)VCF files. This impacts read performance.
The next subsection sheds a bit more light on how VCF-based solutions create the N+1 problem from a data management perspective, and the last subsection explains how TileDB solves this problem with a more principled, database-oriented approach, while providing first-class support for the gVCF standard.
Limitations of VCF files
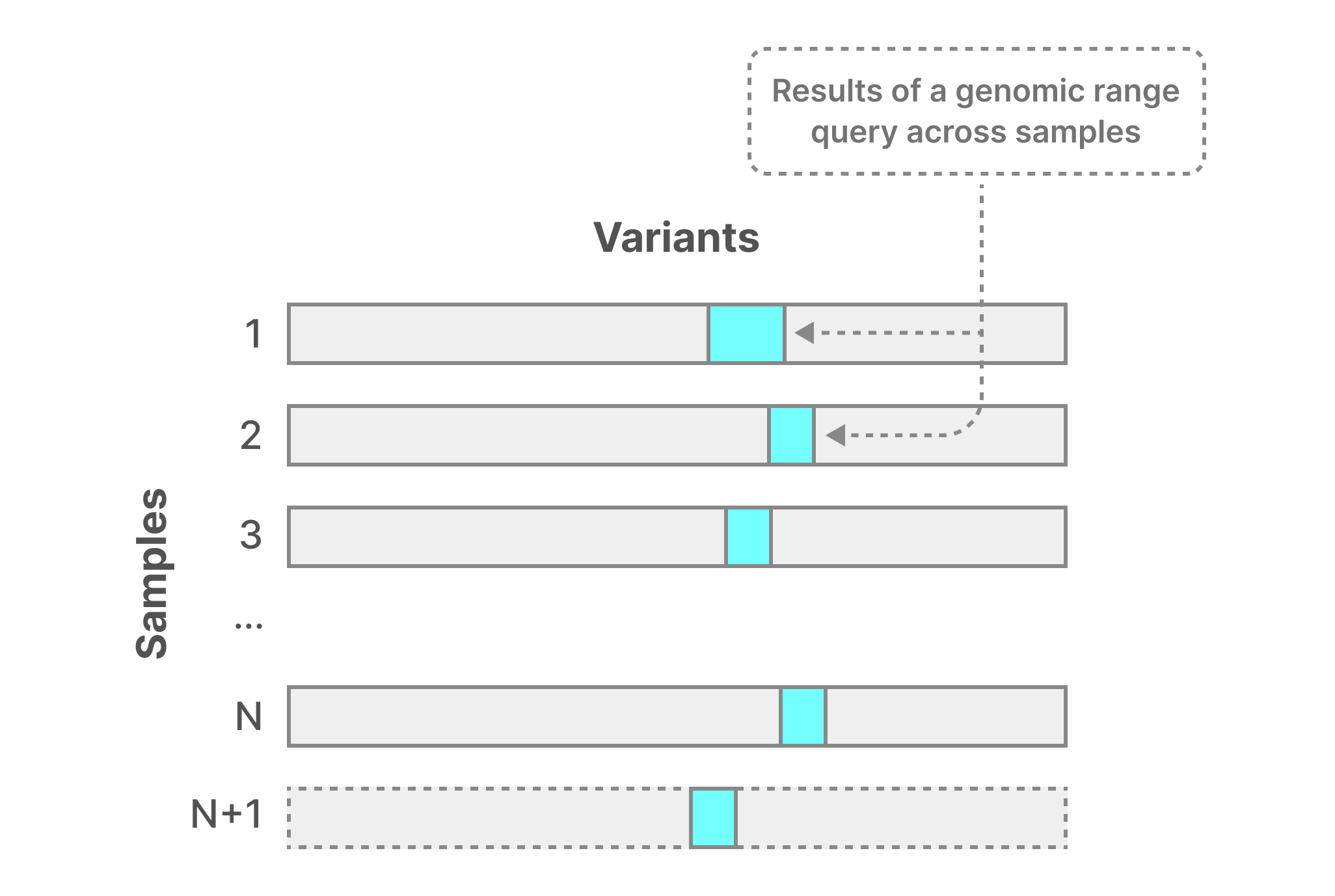
The figure below demonstrates the most basic case, where the user maintains all data as single-sample VCF files. A typical query is one or more genomic ranges across all samples (potentially subsetting on the VCF fields). Although the result for this query may have some locality within each file, a solution performing the query needs to perform N (or N+1) separate range searches across all files. The entire result then has almost no locality, as it is located in very diverse byte ranges in the storage medium. This leads to extremely poor perormance and does not scale well as the number of samples increases.
The second case is to merge all initial N samples into a single file, called CombinedVCF, illustrated in the figure below. That file is optimized for fast genomic range reads across all samples, as this information appears relatively collocated in the same file. This leads to extremely high read performance.
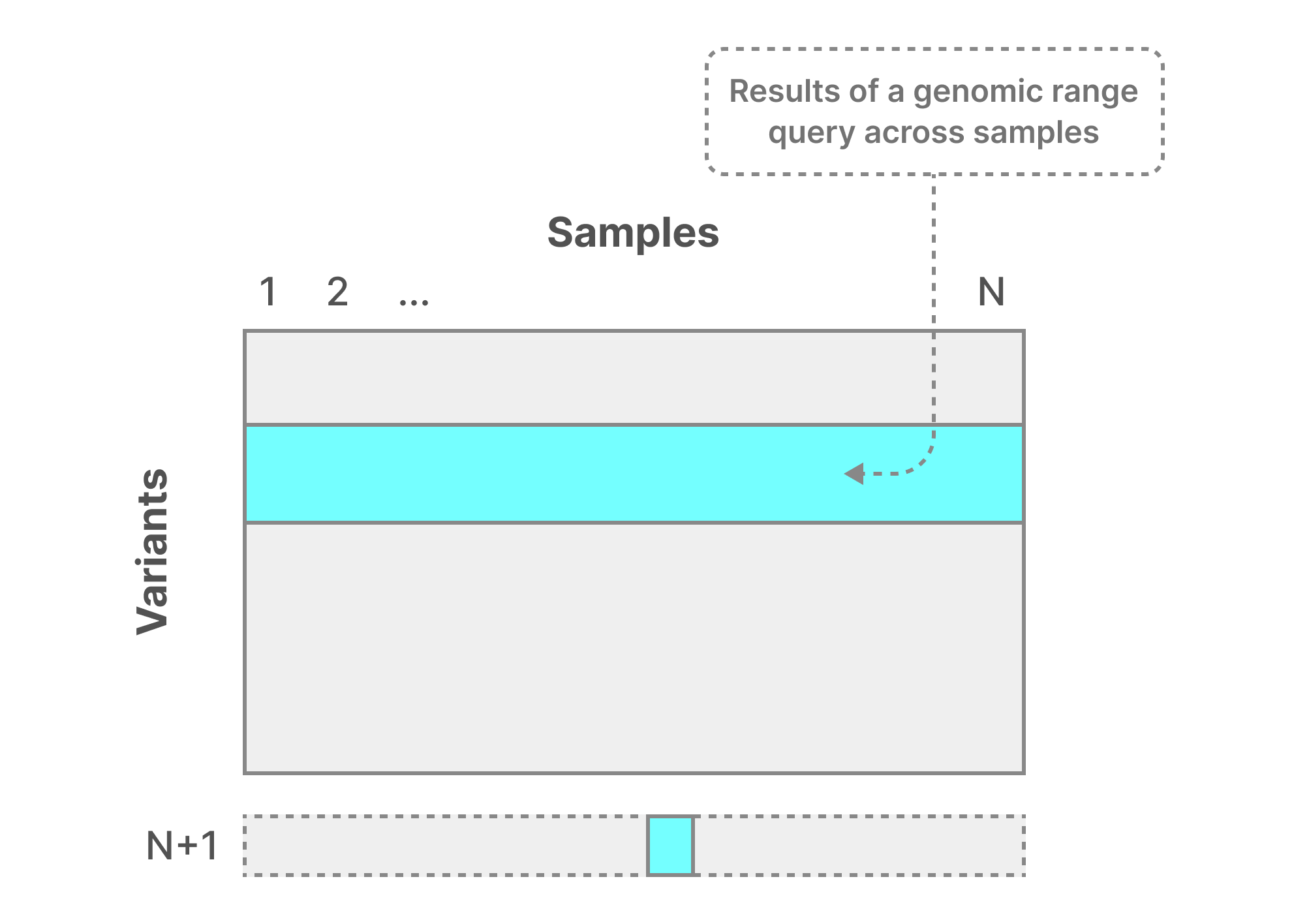
When you wish to add a new sample to a CombinedVCF file, two problems appear:
- The CombinedVCF file needs to be destroyed and completely regenerated. This is because the CombinedVCF file needs to collocate information from each sample in the same genomic range entry. Loosely speaking, this means that the bytes from the new sample file need to be injected in disparate places across the entire CombinedVCF file. For a large value for N, the CombinedVCF file reconstruction cost is prohibitive.
- If the new sample contains a variant that is not present in the CombinedVCF file, a new entry needs to be created, and information from all the N+1 samples needs to be stored in that entry. This leads to a non-linear explosion of the CombinedVCF file size in the number of samples it encompasses.
The above indicates that CombinedVCF-based solutions are not scalable and cannot work for biobank-scale datasets.
How TileDB-VCF solves the N+1 problem
TileDB-VCF adopts a more principled approach to solving this problem. It does so in the following manner:
- It builds upon TileDB arrays, inheriting all the important built-in database functionality, such as efficient storage, indexing and querying, versioning, time traveling, consolidation, and more. TileDB-VCF, coupled with TileDB Cloud, adds further functionality, such as advanced authentication, access control, logging, catalogs, compliance, and more.
- TileDB-VCF offers compressed storage, which scales linearly in the number of samples (eliminating the storage explosion of CombinedVCF). This is because of the way it stores the VCF entries, which is described in detail in the Data Model section.
- TileDB-VCF employs a combination of batch insertions and consolidation to offer an excellent tradeoff between read and ingestion performance. Upon ingestion, the user can specify any number of samples to ingest, and TileDB-VCF can perform the format conversion and write very efficiently, collocating genomic range information across samples (similar to CombinedVCF, but without the storage explosion). When new samples or batches of samples arrive, TileDB-VCF can ingest them without touching the previously ingested samples at all. This further allows embarrassingly parallel ingestion, as TileDB doesn’t need to synchronize anything. Finally, the user can choose to perform an operation called consolidation in the background and without blocking any incoming reads. The consolidation process reorganizes the data, so that it increases the locality of genomic range entries across samples (again, similar to CombinedVCF but without the storage explosion).
TileDB-VCF offers excellent performance for both ingestion and reads, in addition to bringing more management rigor akin to advanced database solutions in other applications.